Some informatiom of Cryo-electron microscopy ( this subjecs & mater wines 2017's nobel prize of Chemestry )
( sources ;- INTERNATE WRITINGS )









( sources internate writings )

Cryo-electron microscopy: A primer for the non-microscopist
What is cryo-EM?
Over the past decade, the phrase “cryo-electron microscopy”, often abbreviated as “cryo-EM”, has evolved to encompass a broad range of experimental methods. At the core, each of these is based upon the principle of imaging radiation-sensitive specimens in a transmission electron microscope under cryogenic conditions. In biology, applications of cryo-EM now span a wide spectrum, ranging from imaging intact tissue sections and plunge-frozen cells to individual bacteria, viruses and protein molecules. Cryo-electron tomography, single-particle cryo-electron microscopy, and electron crystallography are all sub-disciplines of cryo-EM that have been used successfully to analyze biological structures in different contexts. These methods have been used singly as well as in hybrid approaches, where the information from electron microscopy is combined with complementary information obtained using X-ray crystallographic and NMR spectroscopic approaches.
A large number of reviews are published annually highlighting the rapid strides being made in the cryo-EM field [1–14]. These reviews feature detailed methodological advances in specific sub-disciplines such as tomography or single-particle imaging, and report new insights into biological mechanisms revealed through the application of cryo-EM technology. The present review has a different focus and is directed towards students, non-specialists, and researchers in other disciplines who seek a primer on the overall landscape of cryo-EM and an appreciation of the range of biological systems that can be investigated using this technology. Our purpose, therefore, is to use selected examples to provide an introduction to cryo-EM that demystifies the technology and conveys a sense of excitement about this growing area of structural biology.
We begin the review with a brief introduction to imaging using an electron microscope, including the principles underlying data collection and the distinction between cryo-EM and other electron microscope applications that involve imaging under non-cryogenic conditions. In subsequent sections, we highlight different cryo-EM applications that are used to investigate structures ranging in size from cells and viruses to small molecular complexes. We detail the use of cryo-electron tomography to analyze one-of-a-kind entities, such as whole cells, viruses and even molecular complexes, employed with and without the use of sub-volume averaging. Then we introduce single-particle averaging, the process of combining large numbers of 2D projection images to determine 3D structures. And finally, we discuss how averaging methods can be used to determine structures of proteins, at near-atomic resolution, when they are present in ordered assemblies such as two-dimensional crystals. We end with a brief description of the Electron Microscopy Data Bank (EMDB; emdatabank.org), which, together with the Protein Data Bank (PDB; pdb.org), serves as a publicly available repository for EM data at all resolutions.
Imaging with an electron microscope
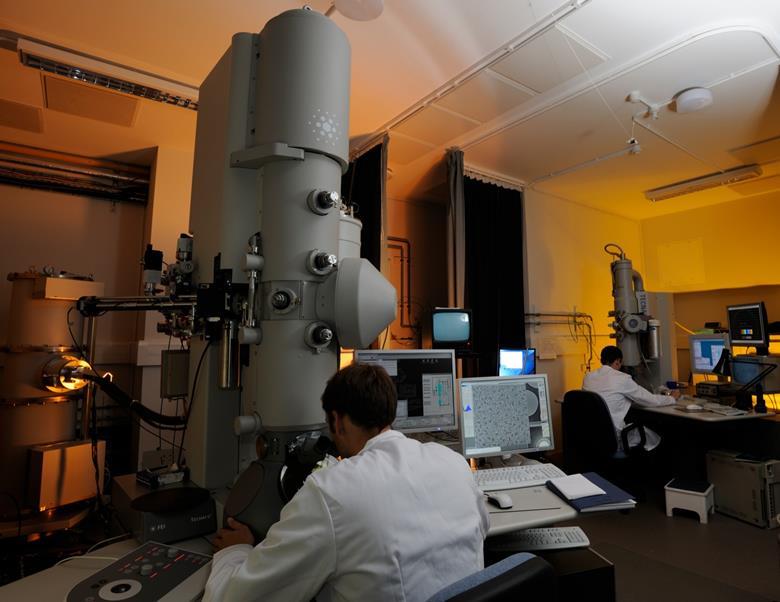
Imaging biological objects in an electron microscope is, in principle, analogous in some respects to light-microscopic imaging of cell and tissue specimens mounted on glass slides. In light microscopy, visible photons serve as the source of radiation; once they pass through the specimen, they are refracted through glass optical lenses to form an image. In electron microscopy, the radiation is electrons, emitted by a source that is housed under a high vacuum, and then accelerated down the microscope column at accelerating voltages of typically 80–300 kV. After passing through the specimen, scattered electrons are focused by the electromagnetic lenses of the microscope (Figure 1a). The most significant difference between optical microscopy and electron microscopy, however, lies in the resolving power of the two methods. Resolution is directly influenced by the wavelength of the imaging radiation source: the shorter the wavelength, the higher the attainable resolution. The resolution possible with visible light (wavelengths ~ 4000–7000 Å; 1 Å = 10−10 m), is significantly less than that achieved with electron sources in a typical transmission electron microscope (wavelength of ~ 0.02 Å for operation at 300 kV). Thus, the wavelength of electron radiation itself does not impose a limit on the resolution that could be obtained when imaging biological macromolecules. The possibility of imaging biological structures with electrons was demonstrated convincingly in 1975 with the determination of the structure of bacteriorhodopsin at ~ 7 Å resolution [15]. These pioneering studies led to the eventual determination of near atomic resolution maps for bacteriorhodopsin [16, 17], and other membrane proteins such as aquaporin [18]. These studies further laid the foundation for recent progress in generation of atomic resolution models for numerous icosahedral viruses [19]. Even higher resolutions have been attained for non-organic samples, which can withstand considerably higher electron doses than biological materials during imaging without loss of structural integrity. For example, individual gold atoms can be directly visualized in high-end electron microscopes from most manufacturers, and the 3D structures of gold nanoparticles have been obtained at atomic resolution using electron tomography [20].
Image formation in the electron microscope. (a) Schematic illustrating image formation in an electron microscope, highlighting the similarities between electron and optical microscopy. (b) Schematic illustrating the principle of data collection for electron ...
So why is it not possible to routinely image individual proteins, viruses and cells in their native state directly in an electron microscope at atomic resolution? The primary reason this is challenging is because of the extensive damage that results from the interaction of electrons with organic matter. Electron irradiation leads to the breaking of chemical bonds and creation of free radicals, which in turn cause further secondary damage [21–23]. One way to mitigate electron-induced sample damage is to use the method of negative staining, in which accessible molecular surfaces are coated with reagents containing heavy atoms, such as uranyl acetate, that are much less radiation-sensitive than organic matter. Because these stains do not penetrate into biological samples, they essentially make a cast of the specimen surface, a high contrast “relief” of the surface, albeit at the expense of internal structural information and with the potential for artifacts such as sample flattening. Nevertheless, this has been a common mode of specimen preparation for many decades in conventional transmission electron microscopy, and has been routinely used for visualization of cells, viruses and proteins, yielding, in the latter case, structures at resolutions of ~ 40 Å to ~ 20 Å [24, 25]. Images of unstained samples or samples that are embedded in sugars such as trehalose, which acts to stabilize proteins in aqueous solution and to increase the image contrast [26], can be obtained at sufficiently low electron doses to yield internal structural information. However, the lower electron doses result in images with a poor signal-to-noise ratio, as unstained biological samples are composed primarily or entirely of low atomic number elements that scatter electrons poorly. Therefore, the difficulty in high-resolution imaging of unstained specimens is that electron doses that are low enough to minimize radiation damage and preserve the specimen generate noisy images, while electron doses that are high enough to get a good signal-to-noise ratio lead to unacceptable levels of specimen damage.
Two approaches are used in modern “high-resolution” electron microscopy to solve this problem. The first approach involves “cryo-electron microscopy”, in which imaging is performed using frozen specimens maintained at either liquid nitrogen or liquid helium temperatures. Almost four decades ago, measurements on the decay of diffraction intensities at room temperature versus cryogenic temperatures established that cryo-electron microscopy could reduce the effects of radiation damage [22]. The development of methods to rapidly freeze (vitrify) biological specimens in a layer of glass-like ice [27, 28], which could then be imaged at liquid nitrogen and/or helium temperatures, paved the way for this method to become commonly used [10, 29]. Plunge-freezing aqueous solutions into a cryogen, such as liquid ethane cooled by liquid nitrogen, is a method used to prepare specimens for cryo-EM applications in tomography, single-particle imaging and for helical and two-dimensional crystals. Imaging at liquid nitrogen temperatures reduces the extent of radiation damage by as much as 6-fold compared to ambient temperatures [28]. This means that, for the same amount of radiation damage, an image can be recorded at cryogenic temperatures with a much higher electron dose [30, 31]. Both liquid nitrogen and liquid helium have been used successfully to obtain 3D reconstructions at near-atomic resolution; a careful comparison of the two cryogens indicate that they show similar protective effects in resolutions between ~ 4–20 Å [32], although the choice of cryogen has long been under debate [28, 33]. Another way to increase the signal-to-noise ratio involves averaging images of a large number of identical units, in an approach that has an intellectual connection to the way in which scattering of X-rays by billions of molecules is averaged to obtain structural information in X-ray crystallography. This technique was first used in the context of helical assemblies [30] and two-dimensional protein crystals imaged at room temperature [31], as well as under cryogenic conditions [27]. Single-particle imaging and electron tomography of individual molecular complexes also invoke the principle of averaging multiple images collected from frozen-hydrated specimens of molecular complexes. These two ideas, namely the concept of imaging at cryogenic temperatures and the concept of averaging multiple low-dose images, form the basis of modern high-resolution biological electron microscopy. The application of these principles to biological imaging is discussed in the following sections, although we will note instances where they are not always used in concert.
Cryo-electron tomography: cellular and sub-cellular structures
One approach to obtaining 3D structures of macromolecular assemblies using electron microscopy is tomography [1, 34, 35], in which a series of images is collected, with each image taken at a different tilt relative to the direction of the incident electron beam (Figure 1b). For imaging thicker specimens such as large viruses or whole bacterial or eukaryotic cells, images are generally collected with the aid of an imaging filter that improves contrast by eliminating inelastically scattered electrons. These images can then be combined computationally, using a strategy similar to that used in computerized axial tomography (CAT), to generate “tomograms”, essentially 3D images of the specimen. Two general types of strategies exist for the further analysis of tomographic data. When specific molecular entities can be clearly identfied within cells or on the surface of viruses, or when molecular complexes are imaged in vitro, averaging methods can be employed to obtain more detailed structural information than is present in the original tomograms. However, many cellular sub-structures (or organelles in the case of mammalian cells) are morphologically heterogeneous, necessitating comparison across multiple tomograms to identify patterns in structural variation.
Electron tomographic imaging requires that specimens be thin enough for the incident electron beam to be transmitted, typically not much more than ~ 0.5 microns even for microscopes capable of operation at 300 kV. Accelerating voltages of 200–300 kV enable greater sample penetrance of the electron beam [34] than microscopes operating at lower voltages. Samples that are sufficiently thin can be negatively stained for room temperature tomography or plunge-frozen under native conditions for cryo-electron microscopy. Thicker samples, such as tissues or cell pellets, require more pretreatment [13]. Classically, samples destined for room temperature tomography are chemically fixed, dehydrated with sequential graded steps of solvent exchange, plastic-embedded and cured to form a block. The block is cut into sections using a diamond knife, and the sections are treated with stains such as uranyl acetate or lead citrate to generate contrast. Thicker samples can also be high pressure-frozen or slam-frozen to promote formation of vitreous, unstructured ice, rather than crystalline ice, which disrupts native cellular architecture. Frozen samples then undergo freeze-substitution for dehydration and fixation, followed by low-temperature plastic embedding, thin sectioning and staining, as above. When necessary, samples of bacterial cells [36] or mammalian cells [37, 38] can also be thinned under cryogenic conditions using focused ion beam milling. Fortunately, many bacteria have thicknesses around 0.5 μm, thus reducing the need for significant sample manipulation. This has made bacterial tomography a particularly productive area for the application of cryo-electron tomography (reviewed in [3]).
Numerous insights have emerged from these tomographic studies, such as the organization of the bacterial cytoskeleton [13, 39], flagellar motor [40], lateral arrangement of membrane protein assemblies [41], visualization of DNA ejection in Bacillus anthracis spore-binding phage [42] and of cell-cell fusion [43], to name just a few examples. Another illustration of the power of tomographic imaging in unraveling internal bacterial structures is the recent discovery of the spiral organization of the bacterial nucleoid [44] in Bdellovibrio bacteriovorus cells (shown in Figure 2). In addition to revealing the twisted nucleoid structure, the tomograms also reveal the localization of ribosomes at the interface between the nucleoid and the cytosol, as well as the ordered arrangement of the chemoreceptor assemblies in the inner membrane.
Use of cryo-electron tomography to image the interior architecture of intact bacterial cells. (a,b) Illustration of spiral architecture of the nucleoid in Bdellovibrio bacteriovorus showing (a) a 210 Å thick tomographic slice through the 3D volume ...
Other applications of cryo-electron tomography have included imaging of thin regions of mammalian cells [45] and investigation of a variety of filamentous and cytoskeletal assemblies [46–48] and of thin vitreous sections prepared from mammalian cells and tissues [38, 49], leading to significant new information on sub-cellular structures [1, 8, 50]. As cellular cryo-electron tomography progresses towards charting macromolecular landscapes [51–54], tomography of vitreous cryo-sections (reviewed in [55]) is emerging as a particularly important tool for the investigation of large cells and tissues. Historically, this technique has been hindered by problems with creating proper attachment of the ribbons of vitreous sections to the support film of the electron microscopic grid. Recently, electrostatic charging has been used to successfully attach vitreous sections to the EM support film [56]. Figure 3 shows a relatively long ribbon of vitreous cryo-sections pulled from the edge of the microtome blade over the EM grid (Figure 3a-c). It is then attached using electrostatic charge to the EM support film. S. cerevisiae cells can be seen scattered throughout the vitreous section, shown at higher magnification (Figure 3e). Another major limitation has been artifacts that are introduced during sectioning, evident in most images [57, 58], although the structures of large macromolecular complexes such as 80S ribosomes remain unaltered by section-induced compression (Figure 3g) [49]. Nevertheless, electron diffraction patterns from high pressure-frozen lysozyme crystals subjected to vitreous cryo-sectioning can extend to 7.9 Å [59], and there is hope that the throughput of many of the steps can be improved, suggesting that vitreous cryo-sectioning could become a routine method for observing macromolecular structures in their native cellular environment.
Imaging of vitreous sections by cryo-electron microscopy. (a) An EM grid is placed at the diamond knife-edge using tweezers with a bent tip. (b) A ribbon of vitreous sections (white arrowheads) is guided over the EM grid using an eyelash (white arrow) ...
As discussed above, tomography is emerging as a powerful method to visualize one-of-a-kind, structurally heterogenous entities at resolutions between ~ 100 Å and ~ 50 Å. These resolutions are two orders of magnitude better than can be achieved by visualizing subcellular detail using optical microscopy, filling a critical gap in cell biology. In addition, cryo-electron tomography is a unique approach for imaging all of the cellular components simultaneously in unstained cells, as the image contrast arises from the intrinsic scattering of electrons by the protein and nucleic acid components. The development of clonable labels for use in electron microscopy [60, 61], as well as of correlative electron-microscopic/light-microscopic techniques [62, 63], provides new avenues to determine the localization of individual biological complexes in situ in cells when specific labeling is desired. While the attainable spatial resolution of fluorescently-labeled proteins within a cell can reach resolutions better than ~1000 Å, unlabeled cellular protein complexes are not visualized by light microscopy. Identification of fluorescently labeled entities at low resolution combined with correlative electron microscopy could thus be highly useful for describing subcellular architecture, while simultaneously targeting specific molecules of interest.
While cryo-electron tomography provides a unique opportunity to explore the organization of protein complexes and cellular components in situ in intact viruses, cells and tissues, individual tomograms from protein complexes are at resolutions that are often not useful for describing molecular structures. However, in cases where there are multiple copies of the structure of interest, it is possible to improve the final resolution by averaging. This procedure, in which multiple copies of sub-volumes containing the assembly of interest are extracted from tomograms, aligned, classified and averaged in 3D, is emerging as a powerful approach at the interface between structural and cell biology [35, 64–69].
One area in which sub-volume averaging has been particularly useful is in structural studies of viral surface proteins, where crystallographic structural information can be difficult to obtain from proteins in their native state. For example, sub-volume averaging approaches have yielded numerous 3D structures, at ~ 20 Å resolution, of trimeric HIV (human immunodeficiency virus) and SIV (simian immunodeficiency virus) envelope glycoproteins in complex with neutralizing antibodies (Figure 4a, 4b) [70–73], providing new and fundamental insights into the mechanism of viral entry. Sub-volume averaging can also be applied to soluble protein complexes where there is excellent contrast between the protein assembly and its surrounding milieu (Figure 4c, 4d) [35, 74, 75]. Subvolume averaging has also led to structural information on complex systems as yet intractable to X-ray crystallography; examples of this include bacterial membrane protein assemblies [76, 77] and myosin V assemblies [78]. The resolutions that have been achieved with this approach are typically in the range of ~ 30 Å to ~ 20 Å. When unambiguous fitting constraints are available, such as the presence of one or more antibody Fab fragments bound to specific sites, individual sub-components whose structures have been determined by X-ray crystallography and NMR can be fitted reliably into the tomographic density maps. The accuracy of this type of fitting is only likely to get better with improvement in the resolutions obtained with tomographic density maps [79] using methods such as “constrained single-particle tomography” that have been developed recently [80]. In this approach, the accuracy of determining molecular orientations is greatly improved by exploiting the known angular constraints present in tomographic tilt series. The finding that resolutions as high as ~ 8 Å can be obtained with this strategy for averaging and 3D reconstruction is thus an exciting development that could have broad applicability for structural analysis of molecular conplexes using cryo-electron tomography.
Structural analysis of membrane protein complexes using cryo-electron tomography combined with sub-volume averaging. (a) Tomographic slice through a field of HIV recorded from a specimen grid that was plunge-frozen and stored at liquid nitrogen temperatures. ...
Single-particle cryo-electron microscopy
Perhaps the most commonly used variant of cryo-electron microscopy is single-particle analysis. In this technique, data from a large number of 2D projection images, featuring identical copies of a protein complex in different orientations, are combined to generate a 3D reconstruction of the structure. When atomic models are available for some or all of the sub-components of the complex, they can be placed or fitted into the density map to provide pseudo-atomic models, considerably extending the information obtained by electron microscopy (reviewed in [10, 81]).
As in most other cryo-EM applications, the first step in single-particle imaging involves spreading soluble complexes across a hole in a carbon film. The specimen is then plunge-frozen, creating a thin layer of vitreous ice that ideally contains identical copies of the complex present in different orientations. The thickness of the ice layer can vary from a few hundred to a few thousand Ångstroms, and is influenced both by the dimensions of the particle and the composition of the buffer. Starting with images of fields containing many molecular complexes, individual particles are selected by hand or by automated algorithms. Once selected, statistical methods, such as principal component analysis, multivariate analysis or covariance analysis, can be used to sort images based on variations in their structural features. A number of computer programs such as EMAN [82], SPIDER [83] and IMAGIC [84], which have been available for many years, can carry out these strategies for image processing. The degree of relatedness between individual “particle” images is used to identify clusters of similar images within the data set. Related images are then averaged to obtain characteristic projection views of the complex at much higher signal-to-noise ratios than in the original images. Iteration of this classification process, using characteristic views of the newly generated class averages as alignment references, improves the accuracy of alignment and permits visualization of finer structural features.
Generation of a 3D reconstruction from the 2D electron microscopic projection views of the molecular complex is dependent upon knowing the relative orientations of all of the particles. The steps involved are mathematically complex, and take advantage of the central projection theorem, which states that, for a 3D object, the Fourier transform of each 2D projection is a central slice through the 3D Fourier transform of the object (Figure 5). Thus, by acquiring a sufficiently large number of molecular images that encompass a wide range of orientations relative to the electron beam, one can build up the 3D reconstructions one image at a time. The 2D Fourier transform of each image contributes a single slice to the 3D Fourier transform. Relative orientations of a pair of images can be derived from the fact that the Fourier transforms of two projections share a common line where they intersect in 3D Fourier space. Angular assignment by identifying common lines can be conducted in Fourier space, as typified by the methodology developed originally for icosahedral virus reconstructions [85], or in real space by obtaining a set of 1D line projections from each pair of 2D class average images and assigning relative orientations based on identifying the line common to both sets [86]. Pair-wise comparisons of at least three class averages to generate three common lines is necessary to assign the relative angular orientations of each projection. Following angular determination, an initial 3D reconstruction can be obtained using real-space reconstruction methods or Fourier-space-based methods. Many variants of these general strategies exist, including those tailored for highly symmetric samples or those with helical symmetry [10, 87]. Initial maps can be refined to higher resolution using projection-matching refinement strategies, which compare individual particle images to uniformly sampled re-projections of the 3D map, performed in real space (using programs such as IMAGIC or SPIDER) or in Fourier space (using programs such as FREALIGN). If appropriate, available atomic coordinates of corresponding protein components derived from X-ray or NMR analyses can be then fitted into the final structure using programs (such as UCSF Chimera) designed for rigid-body or flexible fitting [10, 88, 89].
Principle of reconstruction of 3D structure by Fourier inversion. Using the “Fourier duck” as the prototypical biological specimen, the schematic illustrates that projection images of the object, each with a different orientation, have ...
Distinct single-particle strategies that are not reliant on common lines approaches are employed to better handle commonly encountered situations such as the presence of preferred molecular orientations on the grid or of multiple molecular conformations/states. This latter problem arises when different conformations of a homogeneous sample, different ligand occupancy of a complex, or different oligomerization states of a complex coexist within the sample, leading to size variability or multiple symmetries. The use of geometric constraints combined with computational approaches is particularly useful in samples where the particles present on the grid display a preferred orientation relative to the incident electron beam. For example, in random conical tilt imaging [90], two images of each grid region are recorded, the first with the specimen stage tilted 50–60 degrees and the second after the stage position is returned to 0 degrees, and the information is combined. This strategy is also useful when there is conformational heterogeneity because the untilted molecular images can be classified into distinct groups of 2D projection images, which can then be used in conjunction with the corresponding tilted images to obtain multiple 3D reconstructions. Although random conical tilt effectively samples a large swath of Fourier space, this approach still results in a conical region of missing data in 3D space (“missing cone”), given the practical limitation to the highest tilt angles that can be used to obtain useful images. A recently proposed orthogonal tilt reconstruction scheme [91] circumvents the missing cone problem by collecting +/− 45 degree tilts of a large population of randomly oriented particles; here the two views of each molecule are related by a 90 degree rotation. One tilt set is sorted to generate class averages while the corresponding orthogonal images of each class average sample an array of equatorial views around a sphere, with specific positions dictated via angular information obtained during the initial alignment and classification step. Initial 3D reconstructions of each class can then be determined and compared to identify regions of conformational heterogeneity.
The highest resolutions achieved using single-particle approaches have been found when imaging icosahedral viruses. Two factors account for this: (1) the presence of high symmetry (60-fold or higher) confers a powerful advantage for averaging within each virion, and (2) the large size of the virions (tens of megadaltons in most instances) provides excellent contrast for accurately determining particle orientations in projection images. Specialized software that is targeted to automatically solving icosahedrally symmetric structures works by taking advantage of the special symmetry features of these viruses [92]. The ordered packing arrangement also lowers conformational heterogeneity of viral protein components. Because of these features, structures of numerous icosahedral viruses have now been determined to resolutions better than 4 Å (reviewed in [11]), and approaching 3 Å in selected cases (Figure 7) [19]. Other recent highlights include reconstructions, at resolutions better than ~ 10 Å, of dynamic protein complexes with much lower levels of symmetry than icosahedral viruses, such as chaperones [93], and complexes with no internal symmetry, such as ribosomes [94]. Notably, several large and symmetric membrane protein complexes have been successfully reconstructed to sub-nanometer resolutions [95, 96], indicating that this is a promising strategy for rapidly obtaining medium-resolution structures of protein complexes.
Determination of structure of a non-enveloped icosahedral virus using cryo-electron microscopy. Visualization of (a) entire structure and (b) selected region demonstrating that near-atomic resolution maps can be obtained for highly ordered assemblies ...
Progress is also being made in technology development for electron microscopy and computational aspects of data processing, leading to extraordinary increases in speed and automation. An example of this is presented in Figure 6, where we show that the 3D structure of GroEL can be determined to a resolution of ~ 7 Å using automated strategies for data acquisition, automated particle selection, and automated 3D reconstruction. In addition to the single-particle methods developed to analyze conformationally heterogeneous specimens [97], experimental and computational strategies that combine full tomographic sub-volume averaging to generate one or more initial 3D reconstructions at low resolution, with single-particle cryo-EM refinement of the structure(s) at higher resolution, can be integrated into a seamless pipeline. Improvements in both computational sorting aspects and increased tomographic resolution [80] promise to make this an exceptionally powerful, streamlined strategy for 3D structure determination.
Automation of macromolecular assembly structure determination by cryo-EM single-particle analysis. (a) Images of a cryo-EM grid at sequentially higher magnification, beginning (left) with an image of the entire grid and concluding with an image of individual ...
Cryo-electron microscopy of ordered assemblies
Because 2D projection images can contain information at very high resolutions, in some cases exceeding 3Å, it is possible, in principle, for structures determined by cryo-electron microscopy to achieve near-atomic resolution [98], even in the absence of high symmetry. However, the intrinsic conformational heterogeneity of most isolated macromolecular assemblies poses a major limitation to reaching this goal [99]. One solution to this challenge is to find experimental conditions that induce the formation of helical or crystalline assemblies of the protein of interest. In cases when this is successful, the ordered packing ensures a high degree of conformational homogeneity, while knowledge of the geometry of the packing arrangement provides a highly accurate determination of the relative alignment of the repeating units. This strategy has been extremely effective with membrane proteins that form two-dimensional crystals in the plane of the membrane, allowing structure determination at resolutions approaching ~ 3 Å, as in the case of bacteriorhodopsin (Figure 8) [16]. In addition to two-dimensional crystals, other types of ordered assemblies such as tubular crystals and helical assemblies have also been extremely valuable for structural analysis of conformational changes at resolutions in the 7 Å to 4 Å range. One of the factors limiting resolution even in these specialized cases is the steep fall-off in image amplitudes at high resolution due to factors such as beam-induced specimen movement, charging from incident electrons, and mechanical instabilities in the microscope. In the case of two-dimensional crystals, this problem can be partially offset with the use of electron diffraction to obtain more accurate amplitudes. When this is possible, it is the best of both worlds, enabling structure determination that combines accurate phases derived from the images with accurate amplitudes derived from the diffraction patterns. Resolutions better than ~ 2 Å have been achieved using electron crystallography [18], making this a powerful approach, although restricted to the few special cases when proteins can be induced to form ordered assemblies such as helical or two-dimensional crystals.
Determination of membrane protein structure using electron crystallography of 2D crystals. (a) Electron diffraction pattern from bacteriorhodopsin crystals, with reflections extending to ~ 2 Å. (b) Structures of bacteriorhodopsin in native and ...
New technology developments and automation
One hurdle that has historically slowed progress in the cryo-EM field has been the time- and personnel-intensive nature of the work. Collecting images for structure determination at the highest resolutions requires precise imaging conditions and specimen placement within the electron microscope. In modern microscopes, the alignments and calibrations involved in imaging tend to be very stable, and are usually performed once for every multi-day session of data collection. However, maintaining and compensating for specimen placement, particularly in single-particle analysis and tomography, presents a significant challenge. The position and orientation of the specimen is usually determined with micrometer precision by a mechanized stage. As the stage is adjusted during data acquisition, the movement of this stage leads to continual small changes in the coordinates of the specimen. This can be compensated for by a combination of beam shift and image shift (for the X and Y axes) and defocus (for the Z axis). Using image and beam shift, several adjacent images can also be collected from a single hole. In tomographic data collection, recording tilt series while maintaining the same region in the field of view can pose a significant obstacle because tilting the specimen holder leads to changes in the Z axis, requiring appropriate adjustment of defocus. These challenges have been addressed successfully with the advent of computerized stages, which together with optically stable microscopes have allowed the development of expert systems capable of managing automated data collection for tomography. Robust data collection packages are now available both as freely distributed (SerialEM [100]) and as commercially available programs (Xplor3D) that effectively automate the tedious task of adjusting image position and defocus for multiple exposures on a single region of interest. Additionally, these programs collect data in a “batch” mode, where the microscopes can continue collecting data from different regions of the specimen for periods exceeding 24 hours without manual intervention.
Automation and improvements in many of the steps in the workflow for cryo-electron microscopy have not only led to dramatic increases in the speed and ease of use, but have also substantially improved data quality. The use of robotics in preparation of frozen-hydrated samples has increased specimen reproducibility and identification of optimal conditions for specimen preparation. Automation in data acquisition, environmental enclosures, the use of patterned grids, and the implementation of remote operation schemes, together with hardware improvements such as automated liquid nitrogen refilling systems, have reduced the need for personnel intervention during data acquisition. The commercial and freely available software routines, which allow automated data collection for single-particle microscopy as well as tomography, have also had a major impact on the development of the cryo-EM field.
One important technical development on the horizon is the use of direct electron detectors [101], which adds to the repertoire of devices currently used (photographic film and charge-coupled device (CCD) cameras) for detection. Two parameters that are important in evaluating detector performance are the modulation transfer function (MTF) and the detective quantum efficiency (DQE). The MTF represents the response of the detector at different spatial frequencies, while the DQE reports on the quality with which incident electrons are recorded at each frequency. Thus the MTF essentially describes the resolution of the detector, while the DQE describes how strong the signal is relative to the noise level. Having the highest possible MTF across the spectrum enables obtaining the best DQE across all frequencies. Although photographic film has remained the standard so far for optimal detector performance, and CCDs have remained the choice for ease of use and automation, the new generation of direct electron detectors appear poised to provide performance that exceeds that of film, while preserving all of the convenience of CCD detectors for automation.
Converting 2D projection images into 3D reconstructions is a process requiring both significant computational power and well-designed image processing algorithms. The broad availability of Graphics Processing Units (GPUs) and large computer clusters over the last decade has been critical for reducing the amount of time required to turn raw data into a 3D reconstruction [102–104]. In addition, image-processing routines that can bypass the need for computationally intensive search strategies for orientation assignment have contributed to additional computational savings [64, 105, 106]. Other important advances in image processing include the development of refinement protocols that require limited user involvement and improved classification strategies for sorting out variability within cryo-EM images [75, 97, 107, 108]. Together, these improvements will enable the analysis of larger and more complex cryo-EM datasets at increasingly higher resolutions.
An emerging paradigm for integrated structural biology
Since the first genomes were published more than a decade ago, genomic and proteomic studies have revealed vast networks of protein-protein and protein-nucleic acid interactions in the cell [109]. This explosion in the genomic and proteomic fields highlights the need for a similar growth in the structural biology field. Already, the application of X-ray crystallographic and NMR spectroscopic approaches have yielded >15,000 unique structures that have been deposited in the Protein Data Bank (PDB). These have included entries from single chain proteins as well as multi-protein complexes, and span the range from small complexes with sizes < 50 kDa that are amenable to analysis by NMR spectroscopy to larger complexes that are amenable to crystallization and analysis by X-ray crystallography, providing a wealth of structural insights into biological mechanisms. However, key gaps remain, especially in the analysis of small, dynamic protein assemblies that are not tractable to analysis by either NMR or X-ray crystallographic methods, but may be uniquely suited for study by cryo-EM. Examples are beginning to emerge where the information from very low resolution cryo-EM maps can nevertheless provide useful information when used in conjunction with NMR spectroscopic information [90].
A glimpse of where the greatest challenges and rewards lie in this emerging discipline can be gleaned by inspection of the data deposited with the EM data bank (EMDB), a world-wide repository for the data generated by all forms of cryo-electron microscopy. EMDB was founded at the European Bioinformatics Institute (EBI) in 2002 and since 2007 has been managed by three partnering entities: the Protein Data Bank in Europe (PDBe), the Research Collaboratory for Structural Bioinformatics (RCSB PDB), and the National Center for Macromolecular Imaging (NCMI). In addition to maintaining the archive and developing services for it, the EMDB plays a key role in leading community-wide initiatives for developing standards and improved practices in the field. EMDB has grown to hold over 1300 released maps, and based on the current growth trend, the holdings are expected to grow 5–10 fold by 2020. A unique bird’seye view of the state of the EM field and current trends can be obtained by analyzing the contents of the EMDB. Two useful tools for this purpose are EMStats (pdbe.org/emstats), which is a web service that presents dynamically generated, up-to-date charts of trends and distributions derived from data mined from the archive, and EMSearch (pdbe.org/emsearch), which is an advanced web search service.
Inspection of the distribution of the released entries belonging to the “single-particle” and “icosahedral” aggregation state categories reveals that icosahedral viruses and ribosomes make up nearly half of the entries, with the rest dominated by assemblies such as chaperones that display a high level of symmetry (Figure 9). Only a quarter of the entries are at resolutions better than 10 Å, which is the resolution at which α-helical regions in the structure can begin to be discerned. Remarkably, the vast majority of these structures at 10 Å or better resolution are from large protein complexes, with only six examples (representing four distinct protein complexes) below 500 kDa, reflecting smaller, potentially dynamic protein complexes (Figure 9). These types of complexes are at the heart of cell function, and understanding how they work will generate fundamental insights that will be key to the development of biomedical therapeutics in the coming decades. It is safe to predict that addressing this critical gap in the landscape of structural biology is where we may expect the greatest future impact of cryo-EM.
Distribution of released entries belonging to the “single-particle” and “icosahedral” categories in the Electron Microscopy Data Bank (emdb.org) as of June 2012. Structures from ribosomes and icosahedral viruses dominate ...
Acknowledgments
Work in our laboratory at NIH is supported by the Center for Cancer Research, National Cancer Institute, NIH, Bethesda, MD. We thank Hong Zhou for permission to use the images presented in Figure 7. More information on our research program is available at http://electron.nci.nih.gov and at
( sources internate writings )
Cryo-electron microscopy

Cryo-electron microscopy (cryo-EM), or electron cryomicroscopy, is a form of transmission electron microscopy(TEM) where the sample is studied at cryogenic temperatures (generally liquid-nitrogen temperatures).[1] Cryo-EM is gaining popularity in structural biology.[2]
The utility of cryoelectron microscopy stems from the fact that it allows the observation of specimens that have not been stained or fixed in any way, showing them in their native environment. This is in contrast to X-ray crystallography, which requires crystallizing the specimen, which can be difficult, and placing them in non-physiological environments, which can occasionally lead to functionally irrelevant conformational changes.
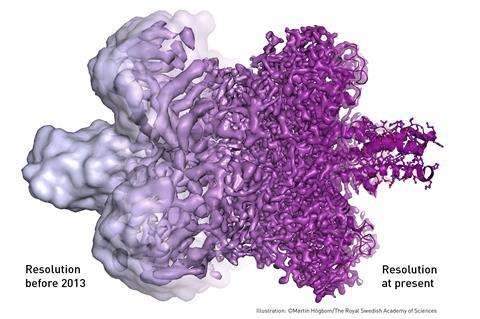
The resolution of cryo-EM maps is improving steadily, and in 2014 some structures at near-atomic resolution had been obtained, including those of viruses, ribosomes, mitochondria, ion channels, and enzyme complexes as small as 170 kDa at a resolution of 4.5 Å.[1] Bridget Carragher and colleagues at the Scripps National Resource for Automated Molecular Microscopy used techniques she and Clint Potter developed to create the first cryo-electron microscopy structural biology image with a resolution finer than 3 Ångströms, thereby elevating cryo-EM as a tool comparable to and potentially superior to traditional x-ray crystallography techniques.[3][4] A 2.2 Å map of a bacterial enzyme beta-galactosidase was published in June 2015.[5] A version of electron cryomicroscopy is cryo-electron tomography (CET), where a 3D reconstruction of a sample is created from tilted 2D images.
Development[edit]
The original rationale for cryoelectron microscopy was as a means to fight radiation damage for biological specimens. The amount of radiation required to collect an image of a specimen in the electron microscope is high enough to be a potential source of specimen damage for delicate structures. In addition, the high vacuum required on the column of an electron microscope makes the environment for the sample quite harsh.
The problem of the vacuum was partially solved by the introduction of negative stains but even with negative stains biological samples are prone to structural collapse upon dehydration of the specimen. Embedding the samples in ice below the sublimation temperature was a possibility that was contemplated early on, but water tends to arrange into a crystalline lattice of lower density upon freezing and this can destroy the structure of anything that is embedded in it.
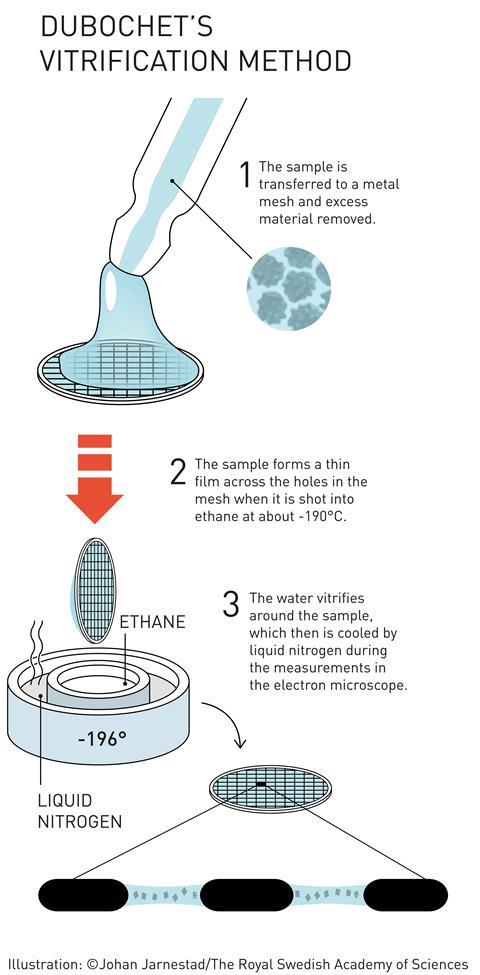
In the early '80s, several groups studying solid state physics were attempting to produce vitreous ice by different means, such as high pressure freezing or flash freezing. In a seminal paper in 1984, the group led by Jacques Dubochet at the European Molecular Biology Laboratory showed images of adenovirusembedded in a vitrified layer of water.[6] This paper is generally considered to mark the origin of cryoelectron microscopy, and the technique has been developed to the point of becoming routine at numerous laboratories throughout the world.
The energy of the electrons used for imaging (80-300 kV) is high enough that covalent bonds can be broken. When imaging specimens vulnerable to radiation damage, it is necessary to limit the electron exposure used to acquire the image. These low exposures require that the images of thousands or even millions of identical frozen molecules be selected, aligned, and averaged to obtain high-resolution maps, using specialized software. A significant improvement in structural features was achieved in 2012 by the introduction of direct electron detectors and better computational algorithms.[1][2]
In 2017, the Nobel Prize in Chemistry was awarded jointly to Jacques Dubochet, Joachim Frank and Richard Henderson, "for developing cryo-electron microscopy for the high-resolution structure determination of biomolecules in solution".[7]
Biological specimens[edit]
Thin film[edit]
The biological material is spread on an electron microscopy grid and is preserved in a frozen-hydrated state by rapid freezing, usually in liquid ethane near liquid nitrogen temperature. By maintaining specimens at liquid nitrogen temperature or colder, they can be introduced into the high-vacuum of the electron microscope column. Most biological specimens are extremely radiosensitive, so they must be imaged with low-dose techniques (usefully, the low temperature of cryo-electron microscopy provides an additional protective factor against radiation damage).
Consequently, the images are extremely noisy. For some biological systems it is possible to average images to increase the signal-to-noise ratio and retrieve high-resolution information about the specimen using the technique known as single particle analysis. This approach in general requires that the things being averaged are identical, although some limited conformational heterogeneity can now be studied (e.g. ribosome). Three-dimensional reconstructions from cryo-EM images of protein complexes and viruses have been solved to sub-nanometer or near-atomic resolution, allowing new insights into the structure and biology of these large assemblies.
Analysis of ordered arrays of protein, such as 2-D crystals of transmembrane proteins or helical arrays of proteins, also allows a kind of averaging which can provide high-resolution information about the specimen. This technique is called electron crystallography.
Vitreous sections[edit]
The thin film method is limited to thin specimens (typically < 500 nm) because the electrons cannot cross thicker samples without multiple scattering events. Thicker specimens can be vitrified by plunge freezing (cryofixation) in ethane (up to tens of μm in thickness) or more commonly by high pressure freezing (up to hundreds of μm). They can then be cut in thin sections (40 to 200 nm thick) with a diamond knife in a cryoultramicrotome at temperatures lower than -135 °C (devitrification temperature). The sections are collected on an electron microscope grid and are imaged in the same manner as specimen vitrified in thin film. This technique is called cryo-electron microscopy of vitreous sections (CEMOVIS) or cryo-electron microscopy of frozen-hydrated sections.
Material specimens[edit]
In addition to allowing vitrified biological samples to be imaged, cryo-EM can also be used to image material specimens that are too volatile in vacuum to image using standard, room temperature electron microscopy. For example, vitrified sections of liquid-solid interfaces can be extracted for analysis by cryo-EM,[8] and sulfur, which is prone to sublimation in the vacuum of electron microscopes, can be stabilized and imaged in cryo-EM.[9]
Techniques[edit]
A variety of techniques can be used in cryoelectron microscopy.[10] Popular techniques include:
- Electron crystallography
- Analysis of two-dimensional crystals
- Analysis of helical filaments or tubes
- Single particle analysis
- Cryo-electron tomography
- MicroED
- Time-resolved Cryo-EM[11][12][13]
The science behind the 2017 Nobel prize in chemistry
The 2017 Nobel prize in chemistry has been awarded to three scientists ‘for developing cryo-electron microscopy for the high-resolution structure determination of biomolecules in solution’.
The laureates are: Jacques Dubochet from the University of Lausanne, Switzerland; Joachim Frank from Columbia University, New York, US; and Richard Henderson from the Medical Research Council Laboratory of Molecular Biology in Cambridge, UK. They each contributed to developing a technique that allows scientists to see the intricate structures of proteins, nucleic acids and other biomolecules, and even study how they move and change as they perform their functions.
What is cryo-EM?
Transmission electron microscopes (TEMs) use a beam of electrons to examine the structures of molecules and materials at the atomic scale. As the beam passes through a very thin sample, it interacts with the molecules, which projects an image of the sample onto the detector (often a charge-couple device; CCD). Because the wavelength of electrons is much shorter than that of light, it can reveal much finer detail than even super-resolution light microscopy (which was awarded the chemistry Nobel prize in 2014).
But some materials – particularly biomolecules – are not compatible with the high-vacuum conditions and intense electron beams used in traditional TEMs. The water that surrounds the molecules evaporates, and the high energy electrons burn and destroy the molecules. Cryo-EM uses frozen samples, gentler electron beams and sophisticated image processing to overcome these problems.
But can’t you look at proteins using x-ray diffraction or NMR?
X-ray diffraction can give very high resolution structures of biomolecules, and several Nobel prizes have been awarded for just that. But to get an x-ray structure, you need to be able to crystallise the molecule. Many proteins won’t crystallise at all. And in some cases, the process of crystallisation alters the structure, so it’s not representative of what the molecule looks like in real life.
Cryo-EM doesn’t require crystals, and it also enables scientists to see how biomolecules move and interact as they perform their functions, which is much more difficult using crystallography.
NMR can also give very detailed structures, and investigate conformational changes or binding of small molecules. But it’s limited to relatively small proteins or parts of proteins, and usually soluble intracellular proteins, rather than those embedded in cell membranes. If you want to study larger proteins, membrane-bound receptors, or complexes of several biomolecules together, cryo-EM is where it’s at.
What did the laureates contribute?
Each of this year’s three winners solved part of the problem of looking at biomolecules in a TEM.
Richard Henderson was the first person to generate an image of a protein using TEM. He packed many copies of the purple light-harvesting protein bacteriorhodopsin into a sample and combined images from multiple angles, using a low-power electron beam, to generate a 3D image of the protein. He continued to refine these techniques over several decades until he could produce images at the same resolution as those from x-ray diffraction.
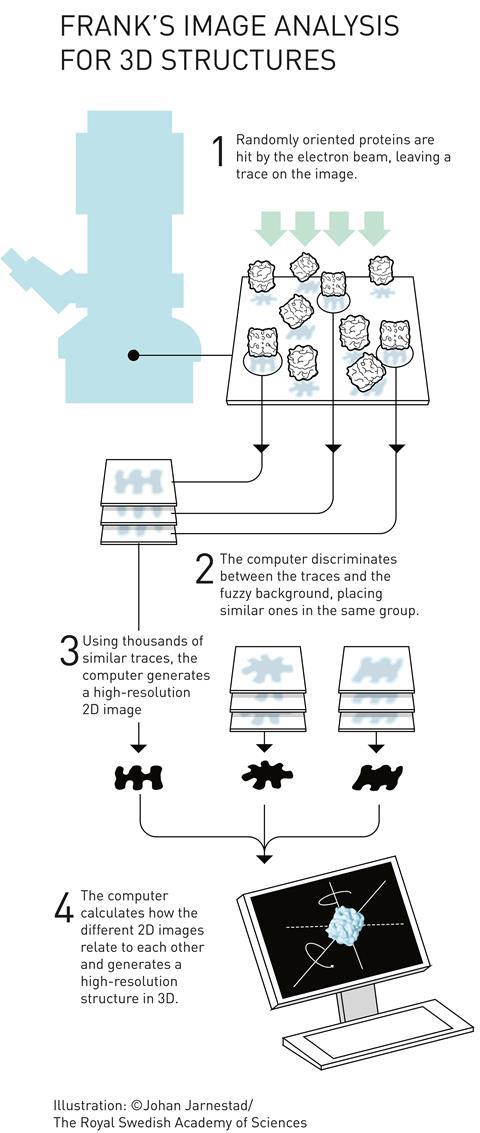
Joachim Frank’s major contribution to the field was in processing and analysing cryo-EM images. He developed computational methods for taking images of multiple, randomly-oriented proteins within a sample and compiling them into sets of similar orientations to obtain sharper 2D images. He could then construct a 3D image from these 2D projections. He used his algorithms to generate images of the ribosome – a massive structure made of several proteins and RNA strands, which is responsible for translating RNA into protein inside cells.
It was James Dubochet who put the ‘cryo’ into cryo-EM. He developed a method for freezing water-based TEM samples so rapidly that the water forms a disordered glass, rather than crystalline ice. This is important because ordered ice crystals would strongly diffract the microscope’s electron beam, obscuring any information about the molecules being studied. Catapulting the sample into a bath of liquid nitrogen-cooled ethane freezes the thin film of water on a TEM sample so quickly that the water molecules don’t have time to arrange into a crystalline lattice. In this ‘vitrified’ sample, the water is disordered but the 3D structure of the biomolecules in the sample is retained. Dubochet created the first images of various viruses using vitrified water samples.
What is cryo-EM used for?
Understanding how biomolecules function and interact is fundamental to biochemistry, and underpins efforts to develop new drugs and medical treatments, and understand and treat diseases.
One recent example is the Zika virus. During the recent outbreak in Brazil, a group of researchers generated a high-resolution 3D image of the virus structure within a few months. This provided a starting point for searching for possible sites that could be targeted by drugs to prevent the spread of the virus.

- PRIVACY
- TERMS OF USE
- ACCESSIBILITY
- ISSUES
- SUBJECTS A-Z
- A-Z OF CONTRIBUTORS
- © CHEMISTRY WORLD 2017
মন্তব্যসমূহ
একটি মন্তব্য পোস্ট করুন